- Home
- Accident & Injury
- Areas We Serve
- Arizona
- Arizona
- Avondale
- Avondale Personal Injury Lawyer
- Avondale Car Accident Lawyer
- Avondale Truck Accident Lawyer
- Avondale Bicycle Accident Lawyer
- Avondale Pedestrian Accident Lawyer
- Avondale Motorcycle Accident Lawyer
- Avondale Rideshare Accident Lawyer
- Avondale Wrongful Death Attorney
- Avondale Dog Bite Lawyer
- Avondale Brain Injury Attorney
- Buckeye
- Buckeye Personal Injury Lawyer
- Buckeye Car Accident Lawyer
- Buckeye Truck Accident Lawyer
- Buckeye Motorcycle Accident Lawyer
- Buckeye Bicycle Accident Lawyer
- Buckeye Pedestrian Accident Lawyer
- Buckeye Rideshare Accident Lawyer
- Buckeye Wrongful Death Attorney
- Buckeye Dog Bite Lawyer
- Buckeye Brain Injury Attorney
- Casa Grande
- Casa Grande Personal Injury Lawyer
- Casa Grande Car Accident Lawyer
- Casa Grande Truck Accident Lawyer
- Casa Grande Motorcycle Accident Lawyer
- Casa Grande Bicycle Accident Lawyer
- Casa Grande Pedestrian Accident Lawyer
- Casa Grande Rideshare Accident Lawyer
- Casa Grande Wrongful Death Attorney
- Casa Grande Brain Injury Attorney
- Casa Grande Dog Bite Lawyer
- Chandler
- Chandler Personal Injury Lawyer
- Chandler Car Accident Lawyer
- Chandler Truck Accident Lawyer
- Chandler Motorcycle Accident Lawyer
- Chandler Bicycle Accident Lawyer
- Chandler Rideshare Accident Lawyer
- Chandler Pedestrian Accident Lawyer
- Chandler Wrongful Death Attorney
- Chandler Dog Bite Lawyer
- Chandler Brain Injury Attorney
- Florence
- Florence Personal Injury Lawyer
- Florence Car Accident Lawyer
- Florence Truck Accident Lawyer
- Florence Motorcycle Accident Lawyer
- Florence Bicycle Accident Lawyer
- Florence Pedestrian Accident Lawyer
- Florence Rideshare Accident Lawyer
- Florence Dog Bite Accident Lawyer
- Florence Brain Injury Attorney
- Florence Wrongful Death Attorney
- Gilbert
- Gilbert Personal Injury Lawyer
- Gilbert Car Accident Lawyer
- Gilbert Truck Accident Lawyer
- Gilbert Motorcycle Accident Lawyer
- Gilbert Bicycle Accident Lawyer
- Gilbert Pedestrian Accident Lawyer
- Gilbert Rideshare Accident Lawyer
- Gilbert Wrongful Death Attorney
- Gilbert Dog Bite Lawyer
- Gilbert Brain Injury Attorney
- Glendale
- Glendale Personal Injury Lawyer
- Glendale Car Accident Lawyer
- Glendale Truck Accident Lawyer
- Glendale Motorcycle Accident Lawyer
- Glendale Bicycle Accident Lawyer
- Glendale Pedestrian Accident Lawyer
- Glendale Rideshare Accident Lawyer
- Glendale Wrongful Death Attorney
- Glendale Dog Bite Lawyer
- Glendale Brain Injury Attorney
- Goodyear
- Goodyear Personal Injury Lawyer
- Goodyear Car Accident Lawyer
- Goodyear Truck Accident Lawyer
- Goodyear Motorcycle Accident Lawyer
- Goodyear Bicycle Accident Lawyer
- Goodyear Rideshare Accident Lawyer
- Goodyear Pedestrian Accident Lawyer
- Goodyear Wrongful Death Attorney
- Goodyear Brain Injury Attorney
- Goodyear Dog Bite Lawyer
- Mesa
- Peoria
- Phoenix
- Phoenix Personal Injury Lawyer
- Phoenix Car Accident Lawyer
- Phoenix Truck Accident Lawyer
- Phoenix Motorcycle Accident Lawyer
- Phoenix Bicycle Accident Lawyer
- Phoenix Pedestrian Accident Lawyer
- Phoenix Rideshare Accident Lawyer
- Phoenix Wrongful Death Attorney
- Phoenix Dog Bite Lawyer
- Phoenix Brain Injury Attorney
- Scottsdale
- Scottsdale Personal Injury Lawyer
- Scottsdale Car Accident Lawyer
- Scottsdale Truck Accident Lawyer
- Scottsdale Motorcycle Accident Lawyer
- Scottsdale Bicycle Accident Lawyer
- Scottsdale Pedestrian Accident Lawyer
- Scottsdale Rideshare Accident Lawyer
- Scottsdale Wrongful Death Attorney
- Scottsdale Brain Injury Attorney
- Scottsdale Dog Bite Lawyer
- Surprise
- Surprise Personal Injury Lawyer
- Surprise Car Accident Lawyer
- Surprise Truck Accident Lawyer
- Surprise Motorcycle Accident Lawyer
- Surprise Bicycle Accident Lawyer
- Surprise Pedestrian Accident Lawyer
- Surprise Rideshare Accident Lawyer
- Surprise Wrongful Death Attorney
- Surprise Dog Bite Lawyer
- Surprise Brain Injury Attorney
- Tempe
- Tucson
- Yuma
- California
- San Diego
- Carlsbad
- Chula Vista
- Coronado
- EI Cajon and La Mesa
- Escondido and San Marcos
- Fallbrook
- Hillcrest
- Lakeside
- Lemon Grove
- Oceanside
- Pacific Beach & Mission Beach
- Poway
- Santee
- Washington
- Yakima
- Vancouver
- Vancouver Personal Injury Lawyer
- Vancouver Car Accident Lawyer
- Vancouver Brain Injury Attorney
- Vancouver Truck Accident Lawyer
- Vancouver Motorcycle Accident Lawyer
- Vancouver Bicycle Accident Lawyer
- Vancouver Pedestrian Accident Lawyer
- Vancouver Rideshare Accident Lawyer
- Vancouver Wrongful Death Attorney
- Vancouver Dog Bite Lawyer
- Washington State
- Washington State Personal Injury
- Washington State Car Accident Lawyer
- Washington State Brain Injury Attorney
- Washington State Truck Accident Lawyer
- Washington State Motorcycle Accident Lawyer
- Washington State Bicycle Accident Lawyer
- Washington State Pedestrian Accident Lawyer
- Washington State Rideshare Accident Lawyer
- Washington State Wrongful Death Attorney
- Washington State Dog Bite Lawyer
- Spokane Valley
- Personal Injury in Spokane Valley
- Spokane Valley Car Accident Lawyer
- Spokane Valley Brain Injury Attorney
- Spokane Valley Truck Accident Claims
- Spokane Valley Motorcycle Accident Lawyer
- Spokane Valley Bicycle Accident Lawyer
- Spokane Valley Pedestrian Accident Lawyer
- Spokane Valley Rideshare Accident Lawyer
- Spokane Valley Wrongful Death Attorney
- Spokane Valley Dog Bite Lawyer
- Tacoma
- Spokane
- Spokane Personal Injury Lawyer
- Car Accident Lawyer in Spokane
- Spokane Brain Injury Attorney
- Spokane Truck Accident Lawyer
- Spokane Motorcycle Accident Lawyer
- Spokane Bicycle Accident Lawyer
- Spokane Rideshare Accident Lawyer
- Spokane Pedestrian Accident Lawyer
- Spokane Wrongful Death Attorney
- Spokane Dog Bite Lawyer
- Seattle
- Seattle Personal Injury Lawyer
- Seattle Car Accident Lawyer
- Seattle Brain Injury Attorney
- Seattle Truck Accident Lawyer
- Seattle Motorcycle Accident Lawyer
- Seattle Bicycle Accident Lawyer
- Seattle Pedestrian Accident Lawyer
- Seattle Rideshare Accident Lawyer
- Seattle Wrongful Death Attorney
- Seattle Dog Bite Lawyer
- Renton
- Auburn
- Bellingham
- Bellingham Personal Injury Lawyer
- Bellingham Car Accident Lawyer
- Bellingham Brain Injury Attorney
- Bellingham Truck Accident Lawyer
- Bellingham Motorcycle Accident Lawyer
- Bellingham Bicycle Accident Lawyer
- Bellingham Pedestrian Accident Lawyer
- Bellingham Rideshare Accident Lawyer
- Bellingham Wrongful Death Attorney
- Bellingham Dog Bite Lawyer
- Redmond
- Redmond Personal Injury Attorney
- Redmond Brain Injury Attorney
- Redmond Car Accident Lawyer
- Redmond Truck Accident Lawyer
- Redmond Motorcycle Accident Lawyer
- Redmond Bicycle Accident Lawyer
- Redmond Pedestrian Accident Lawyer
- Redmond Rideshare Accident Lawyer
- Redmond Wrongful Death Attorney
- Redmond Dog Bite Lawyer
- Bellevue
- Bellevue Personal Injury Lawyer
- Bellevue Car Accident Lawyer
- Bellevue Brain Injury Attorney
- Bellevue Truck Accident Lawyer
- Bellevue Bicycle Accident Lawyer
- Bellevue Motorcycle Accident Lawyer
- Bellevue Pedestrian Accident Lawyer
- Bellevue Rideshare Accident Lawyer
- Bellevue Wrongful Death Attorney
- Bellevue Dog Bite Lawyer
- Kent
- Federal Way
- Federal Way Personal Injury Lawyer
- Federal Way Car Accident Lawyer
- Federal Way Brain Injury Attorney
- Federal Way Truck Accident Lawyer
- Federal Way Motorcycle Accident Lawyer
- Federal Way Bicycle Accident Lawyer
- Federal Way Pedestrian Accident Lawyer
- Federal Way Rideshare Accident Lawyer
- Federal Way Wrongful Death Attorney
- Federal Way Dog Bite Lawyer
- Kirkland
- Kirkland Personal Injury Lawyer
- Kirkland Car Accident Lawyer
- Kirkland Brain Injury Lawyer
- Kirkland Truck Accident Lawyer
- Kirkland Motorcycle Accident Lawyer
- Kirkland Bicycle Accident Lawyer
- Kirkland Pedestrian Accident Lawyer
- Kirkland Rideshare Accident Lawyer
- Kirkland Wrongful Death Attorney
- Kirkland Dog Bite Lawyer
- Everett
- Arizona
- Defective Drugs
- Defective Products
- Other Case Types
- Blog
- Contact Us
Hablamos Espanol - Call 24/7 1 (800) 214-1010
FDA Alert for Invokana Increased Risk of Leg and Foot Amputations
May 18, 2016 — San Diego, CA. ISSUE: FDA is alerting the public about interim safety results from an ongoing clinical trial that found an increase in leg and foot amputations, mostly affecting the toes, in patients treated with the diabetes medicine canagliflozin (Invokana, Invokamet). FDA has not determined whether canagliflozin increases the risk of leg and foot amputations. FDA is currently investigating this new safety issue and will update the public when we have more information.
See the FDA Drug Safety Communication for additional details regarding the ongoing Canagliflozin Cardiovascular Assessment Study (CANVAS) clinical trial.
BACKGROUND: Canagliflozin is a prescription medicine used with diet and exercise to lower blood sugar in adults with type 2 diabetes. It belongs to a class of drugs called sodium-glucose cotransporter-2 (SGLT2) inhibitors. Canagliflozin lowers blood sugar by causing the kidneys to remove sugar from the body through the urine. It is available as a single-ingredient product under the brand name Invokana and also in combination with the diabetes medicine metformin under the brand name Invokamet.
——End——
Judge maintains $2.5 million Risperdal verdict
May 12, 2016 — San Diego, CA. A Philadelphia judge refused to throw out a $2.5 million verdict against Johnson & Johnson’s pharmaceutical unit over allegations its antipsychotic drug Risperdal caused an autistic boy to grow breasts.
As reported by Law 360, Johnson & Johnson subsidiary Janssen Pharmaceuticals argued the $2.5 million verdict was excessive and that a substitution of the plaintiff’s causation expert during the bellwether trial had left the defendant’s counsel unprepared. Philadelphia County Court of Common Pleas Judge Ramy I. Djerassi rejected the company’s bid in a May 4 order.
The family of plaintiff Austin Pledger, now 20, sued Janssen Pharmaceuticals in 2012 alleging the drugmaker’s antipsychotic medication caused him to grow large breasts, a condition known as gynecomastia. Pledger reportedly started taking Risperdal beginning in 2002 when he was just seven years old.
The $2.5 million verdict was handed down by a jury in February 2015; it would be the first of three decisions in connection with Risperdal-related male breast growth made in favor of the plaintiffs. A Maryland man who took Risperdal beginning at nine years old was awarded $1.75 million (later reduced to $680,000) in November 2015, while a Wisconsin man was awarded $500,000 in December 2015.
Janssen did cinch a victory in the second Risperdal case to go to trial in March 2015, when the jury found Janssen negligent in warning about the drug’s risks, but that its negligence did not cause the plaintiff’s abnormal breast growth.
There are currently about 1,500 Risperdal cases pending in a mass tort program in the Philadelphia court.
Risperdal is an antipsychotic drug originally approved by the Food and Drug Administration to treat schizophrenia in adults. Its use was later expanded to treat other mental health disorders in both adults and children, including bipolar disorder and irritability associated with autism spectrum disorder.
Johnson & Johnson shelled out $2.2 billion in 2013 after losing a battle against the federal government, which accused the company of improperly marketing some of its drugs, including Risperdal, for unapproved uses. Learn more on Riperdal lawsuits here: https://www.nationalinjuryhelp.com/defective-drugs/risperdal/
——End——
Latest FDA Recalls and warnings
May 6, 2016 — San Diego, CA. FDA’s latest round up of medical device and drug recalls.
Recalls and Safety Alerts:
Covidien Oridion Labeled Capnostream 20 and Capnostream 20p Patient Monitor Battery Packs by Medtronic: Recall
Recall due to a battery manufacturing defect that may increase the risk of thermal damage in the battery pack…
Fetch 2 Aspiration Catheter by Boston Scientific: Recall
All reports of shaft breakage happened during the procedure, and the broken section was either removed while still partially attached to the catheter shaft or retrieved with a snare, without further patient complications…
Focus Diagnostics Laboratory Examination Kits: Class I Recall
Recall due to poor lamination between the sample reaction wells which could lead to leakage into adjacent wells causing cross-contamination between samples, which could yield false positive, false negative, or invalid test results…
G4 Platinum and G5 Mobile Continuous Glucose Monitoring System Receivers by Dexcom: Class I Recall
The audible alarm may not activate in the receiver piece when low or high glucose levels are detected…
Guardian II Hemostasis Valves by Vascular Solutions, Inc: Recall
The products have been recalled because they pose an increased risk of air leakage that may lead to an air embolism, which could result in serious injury or death…
Urogynecologic Surgical Mesh Implants by Boston Scientific: Notification
FDA is aware of allegations that Boston Scientific’s urogynecologic surgical mesh may contain counterfeit raw material. We are examining these allegations to determine any necessary and appropriate next steps. FDA is not currently aware that the alleged counterfeit raw material contributes to adverse events associated with these products…
Interference between Computed Tomography (CT) and Electronic Medical Devices: Notification
FDA has received a small number of reports of adverse events that are believed to be associated with CT imaging of some implantable and wearable electronic devices…
——End——
Jury awards $55 million to woman in talcum powder lawsuit
May 3, 2016 — San Diego, CA. Johnson & Johnson lost a second battle in court over its talc-based products after a St. Louis jury awarded $55 million to the plaintiff Monday.
Gloria Ristesund, 62, was awarded $5 million in compensatory damages and $50 million in punitive damages. Ristesund was diagnosed with ovarian cancer in 2011 and had used Johnson & Johnson’s talcum powder for 35 years, CNN Money reported Tuesday morning.
Several studies have suggested an increased risk of ovarian cancer in women using talc-based products, such as Johnson & Johnson’s baby powder and Shower to Shower, in the genital region.
During the trial, the plaintiff’s attorney, Allen R. Smith Jr., reportedly told jurors that Johnson & Johnson knew of the possible increased risk of cancer but did not warn consumers. As reported by Law 360, an internal J&J document from 1986 showed retrospective studies implicated talc with an increased risk of ovarian cancer. Those implications were allegedly denied publicly by J&J.
This is the second jury panel to find Johnson & Johnson liable in talcum powder suits this year. The first verdict of $72 million was awarded in February to a woman who passed away from ovarian cancer in 2015.
Johnson & Johnson reportedly said it would appeal Monday’s verdict.
There are currently about 1,200 cases accusing J&J of not adequately warning consumers of the risk of talc-based products pending in Missouri and New Jersey state courts, according to Reuters.
——End——
What are Filshie Clips and are they safe for women?
April 27, 2016 — San Diego,CA. Filshie clips have been used for over 30 years to permanently sterilize women who choose not to have more kids. Filshie clips are used during tubal ligation, a procedure that involves blocking a woman’s fallopian tubes so she cannot get pregnant. The small devices have been used in millions of women worldwide, and now numerous women are stepping forward to report serious side effects related to the clips.
Filshie clips are small, silicone-lined titanium clamps that are placed on the fallopian tubes during tubal ligation surgery. The clips, once closed, create a blockage in the tubes so eggs cannot reach the uterus and become fertilized. Filshie clips are intended to be used as a permanent form of birth control.
Read more here: https://www.nationalinjuryhelp.com/filshie-clips-lawsuits/
——End——
St. Jude Riata leads, devices that regulate heart beats, fail inside patients
April 25, 2016 – San Diego, CA. Medical device manufacturer St. Jude Medical Inc. agreed to pay more than $14 million in settlements more than a year ago for a faulty device that put patients at risk for death.
The company’s Riata defibrillator leads came under scrutiny in 2011 after the FDA initiated a class I recall, its strongest classification, due to the possibility of death or serious injury if the devices failed.
Riata leads are wires that connect implantable cardioverter defibrillators (ICDs) to the heart. ICDs are usually implanted in a person who suffers from a dangerously fast or chaotic heartbeat and helps prevent cardiac arrest by delivering an electric shock if the heart stops beating effectively.
Read more on our blog: https://www.nationalinjuryhelp.com/defective-products/riata-leads-failing/
——End——
Attention Women who have the Essure Birth Control Device.
April 20, 2016– San Diego, CA. The FDA docket for the Essure device closes on May 3, 2016. If you suffered from Essure and haven’t left a comment yet, please take a moment to be heard!
The FDA is asking for your input for the black box warning for Essure.
Be heard and let’s get this device RECALLED!
Here is the link to the docket.
https://www.regulations.gov/#!documentDetail;D=FDA-2016-D-0435-0001
To read about the Essure problems and lawsuits forming please see this page: https://www.nationalinjuryhelp.com/defective-products/essure-contraceptive-implant-lawsuit/
——End——
Does Bard plan to settle pending IVC filter lawsuits?
April 19, 2016 — San Diego, CA. Pharmaceutical company C.R. Bard Inc. could be planning to settle the many lawsuits filed against the company and its inferior vena cava (IVC) filters.
In a 10-k report recently filed with the Securities and Exchange Commission (SEC), Bard hinted that it could plan to settle the nearly 100 lawsuit now pending in district courts across the country, including the U.S. District Court of Arizona.
In the report, which summarizes the company’s financial position, Bard said it “intends to vigorously defend Filter Product Claims that do not settle.” As reported in an article by Mass Tort Nexus, this language could mean the company plans to settle as many lawsuits as possible before they go to trial.
A judge ruled against Bard in 2012 in the first IVC filter lawsuit. The following year, the company settled over 30 IVC filter lawsuits before they went to trial.
Bard could be planning similar settlements for the roughly 95 lawsuits currently pending in district courts. The company also said they were threatened with an additional 130 lawsuits which have not been filed yet.
In light of these suits, Bard said it cannot “give any assurances that the resolution of these claims will not have a material adverse effect on the company’s business, results of operations, financial condition and/or liquidity.”
IVC filters are small, spider-like devices inserted in a patient’s vein just below the kidneys and are intended to catch blood clots before they make their way to the heart or lungs. The devices have come under scrutiny in the past few years because of complications that can arise if they are left in the body for an extended period of time. Parts of the device can break off and perforate the vein or other organs. Sometimes, the entire device breaks free from its original position.
Bard wasn’t the only manufacturer of these potentially dangerous devices. Boston Scientific, Cook Medical and B Braun Medical all had IVC filters on the market at one point.
In the wake of litigation, Bard announced on its website that it no longer markets four of its five IVC filters.
———end———
Medtronic accused of hiding bone graft’s adverse events from the FDA.
April 18, 2016 — San Diego, CA. The largest medical technology company has been called out by a U.S. senator to provide details about injuries associated with its bone graft product.

Medtronic Inc., Minnesota-based manufacturer of a genetically engineered bone grafting product called Infuse, was accused of failing to report adverse events associated with its product to the Food and Drug Administration (FDA) for years.
The Star Tribune reported Sunday, April 10 that Medtronic had discovered roughly 1,000 adverse events from patients who had been injured – and in some cases died – after having Infuse inserted during surgery.
Medtronic said the reports, which were discovered during a retrospective chart review of more than 3,600 patients implanted with Infuse between 2002 and 2006, were “misfiled” for about five years before they were rediscovered in 2013.
Read the story on our blog: https://www.nationalinjuryhelp.com/defective-products/medtronic-infuse-adverse-events/
———–END————–
Federal judge allows Essure suit to proceed – A Victory for Women!
March 29, 2016 – San Diego, CA. Bayer Healthcare suffered defeat this week in federal court, when a judge ruled he would not dismiss a case brought by five women who claim they were harmed by the company’s Essure permanent birth control.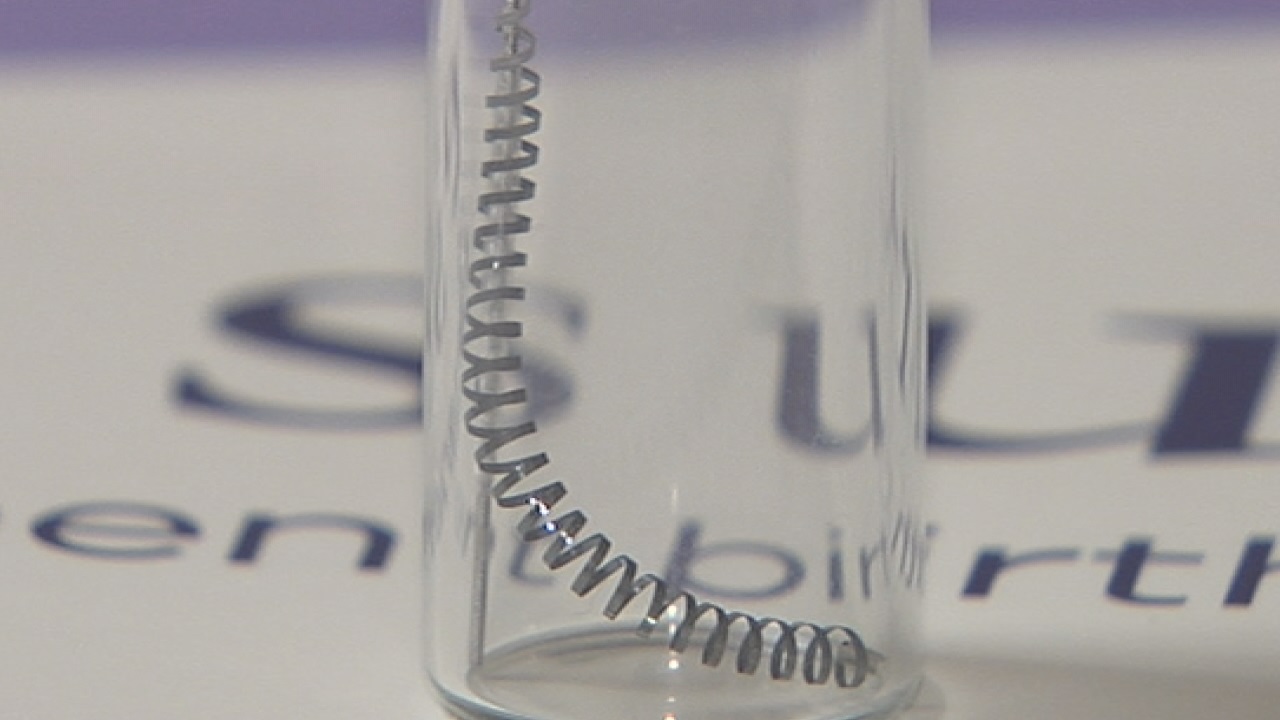
Though U.S. District Judge John R. Padova in Philadelphia rejected 10 of the women’s 12 claims, he is allowing two to move forward, including claims that Bayer used misleading advertising and that it failed to warn about the risks of Essure, reports Philadelphia-based news source philly.com.
Judge Padova is reportedly allowing the plaintiffs to amend their legal arguments to restore another five claims, including one that alleges Bayer’s manufacturing and physician training were negligent.
The decision comes in spite of the U.S. Food and Drug Administration’s (FDA) premarket approval of Essure, which shields the device from product liability litigation in state courts.
Essure came on the market in 2002 after FDA approval and was touted as a less-invasive option for women seeking permanent birth control. Unlike the traditional surgical procedure of “getting your tubes tied,” which involves burning, tying or cutting a woman’s Fallopian tubes to permanently prevent pregnancy, Essure was marketed as a non-surgical procedure that could be done in the doctor’s office.
Essure is made up of two metal coils that are inserted into a woman’s Fallopian tubes by a doctor. Scar tissue is formed around the coils and permanently blocks the tubes, therefore, preventing pregnancy.
Controversy over the safety and efficacy of these devices has sprung up in the past several years, with women speaking out about the harm Essure has allegedly caused them. The potentially harmful side effects reported by women include nickel allergies, pelvic pain, migraines, hair loss, perforation of the Fallopian tubes and migration of the devices into the uterus or abdomen.
And despite a surgery free insertion, many women who want the Essure coils out of their bodies must undergo surgery. Some women may have to have a hysterectomy to get them out.
Despite possible complications, Bayer stands by its product and said in a statement that it was pleased the judge narrowed the case.
“Two prior courts considering similar claims dismissed those suits entirely,” the statement said. “As FDA itself has stated recently, ‘Essure remains an appropriate option for the majority of women seeking a permanent form of birth control.’ Bayer stands by the positive benefit-risk profile of Essure.”
This is one of a number of cases against Bayer and its Essure product. As reported by philly.com, this case is the furthest along in the litigation process and could help shape the legal strategies of other plaintiffs across the country.
—————————-END———————–
J&J settles power morcellator cases; more cash settlements possible
March 22, 2016, San Diego, CA –Johnson & Johnson began settling power morcellator cases Friday — more than a year and half after it issued a worldwide recall of the devices.
As reported by LegalReader, co-lawyer for the plaintiffs Aimee Wagstaff said there are about 100 cases ready to file or that have already been filed in the multi-district litigation (MDL), and Johnson & Johnson has settled several of them.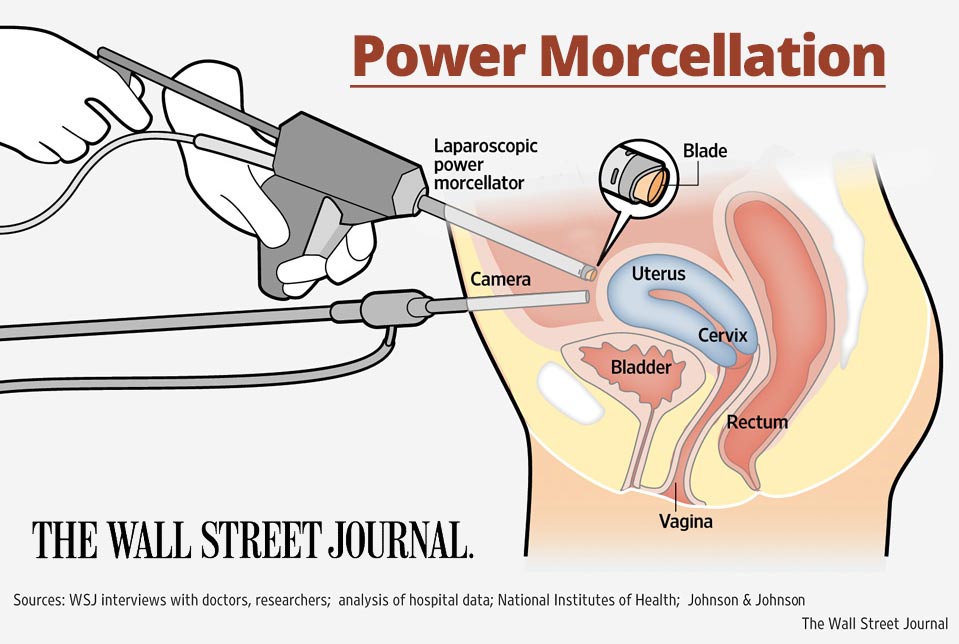
There are still many more power morcellator cases being investigated by attorneys around the country and they are ready to be added to the MDL, as well, Wagstaff told LegalReader.
Power morcellators, manufactured by Johnson & Johnson’s Ethicon division, were often used on women undergoing minimally invasive surgery to treat uterine fibroids. The device broke up tissue into smaller pieces, but in doing so, could spread undiagnosed cancers throughout the women’s abdomens. Once cancer is spread, women’s chances of long-term survival are significantly decreased.
The U.S. Food and Drug Administration (FDA) issued a safety communication in April 2014 warning doctors and patients about the risks of using power morcellators. After analyzing current data, the FDA estimated that about one in 350 women treated for fibroids is found to have unsuspected cancer. There is also no reliable method for predicting or testing whether a woman with fibroids could have uterine cancer, the FDA said.
Ethicon suspended sales of the product in April 2014, the same month the FDA issued its warning, and the first lawsuit against the company regarding power morcellators was filed in May 2014, reported LegalReader.
After the FDA’s Obstetrics and Gynecology Devices Panel met in July 2014 to discuss potential risks of power morcellators, Ethicon issued a worldwide voluntary recall of its product.
A year and eight months later, the company is settling cases with patients who were injured by the popular device.
No comment was issued by either Ethicon or Johnson & Johnson when news of the settlements broke Friday.
————END————–