 Dec. 16, 2016 – San Diego, CA – On Tuesday, President Barack Obama signed into law the 21st Century Cures Act, a bill that was passed by the Senate with overwhelming bipartisan support last week and by the House of Representatives with equal fervor just a week prior.
Dec. 16, 2016 – San Diego, CA – On Tuesday, President Barack Obama signed into law the 21st Century Cures Act, a bill that was passed by the Senate with overwhelming bipartisan support last week and by the House of Representatives with equal fervor just a week prior.
The 21st Century Cures Act had been working its way through Congress since at least 2015 and took on many forms before the one passed by the legislatures this month.
The bill is seen as a bipartisan victory and a means to bring the U.S. healthcare system, well, into the 21st century. It provides a huge $5 billion boost to the budget-strapped National Institutes of Health (NIH) for valuable research and important healthcare initiatives, including Vice President Joe Biden’s Cancer “Moonshot” and President Obama’s Precision Medicine Initiative.
The bill also provides $1 billion in crucial funding for states to combat the opioid epidemic and a necessary boost for mental health care.

Preview of the 21st Century Cures Act
The Cures Act has been touted by supporters as an “innovative game-changer” and a “once-in-a-generation” chance to modernize the healthcare system.
Part of the act’s main mission is to speed the approval process of life-saving drugs and devices to patients suffering from rare diseases.
This comes in the form of billions of dollars in funding for NIH and U.S. Food and Drug Administration.
But critics of the bill say the $5-billion-plus in funding is a smoke screen that distracts from what the Cures Act will really do: relax essential FDA requirements and strengthen the hold Big Pharma already has on the U.S. healthcare system.
Sen. Elizabeth Warren, D-Mass., who was one of only 5 senators to vote against the bill last week, said the 21st Century Cures Act had been “hijacked” by the pharmaceutical industry, as reported by STAT news.
Sen. Warren, like the other few dissenting voices in Congress, said the bill weakens the FDA’s approval requirements for drugs and medical devices in exchange for NIH research funding that might not exist because it must be approved by future legislatures on an annual basis.
“I cannot vote for this bill,” Sen. Warren is reported saying from the Senate floor just days before the House voted for the current version on Nov. 30. “I will fight it because I know the difference between compromise and extortion.”
More than 1,400 lobbyists put pressure on lawmakers to either pass or prevent the 21st Century Cures Act, according to a report by CBS News. Most of those lobbyists represented pharmaceutical companies and their work has amounted to what L.A. Times columnist Michael Hiltzik called a “deregulatory giveaway” to Big Pharma disguised as a triumph for innovation and modern research.
This should not come as a surprise, considering the Cures Act was born of the notion that the Food and Drug Administration was a “bottleneck” to drug and medical device approval and that its vast system of regulations stymied innovation.
The Cures Act is an effort to combat this obstruction and take the U.S. healthcare system forward, but in doing so, it effectively erodes critical regulations put in place at the FDA to ensure patient safety.
This includes loosening the requirements needed to get drugs approved for new uses; expediting the approval of medical devices that fall under a brand-new, vague “breakthrough device” category; and allowing pharmaceutical companies to promote their drugs for “off-label” uses to insurance companies. This is just a sampling.
Real-world evidence replaces the ‘gold standard’
 Under the 21st Century Cures Act, pharmaceutical companies could obtain FDA approval for new uses of existing drugs without having to conduct randomized, controlled trials – considered the “gold standard” of clinical trials. Instead, companies could submit real-world evidence to support their drug’s approval.
Under the 21st Century Cures Act, pharmaceutical companies could obtain FDA approval for new uses of existing drugs without having to conduct randomized, controlled trials – considered the “gold standard” of clinical trials. Instead, companies could submit real-world evidence to support their drug’s approval.
The same goes for satisfying postapproval study requirements, which companies must often conduct after gaining FDA approval to show their medical devices continue to be safe and effective.
What is real-world evidence? According to the Cures Act, real-world evidence is data derived from sources other than randomized clinical trials, including ongoing safety surveillance, observational studies, registries, claims, and patient-centered outcomes research activities.
Although real-world evidence is an important facet of ensuring the safety and effectiveness of drugs and medical devices, nothing should replace the gold standard when it comes to approving these new products and new uses.
Researchers at Science-Based Medicine have raised the alarm bells on this particular provision.
Dr. David H. Gorski, surgical oncologist at the Barbara Ann Karmanos Cancer Institute and managing editor at Science-Based Medicine, wrote in an op-ed that drug companies would “love this provision” because it allows them to skip these large clinical trials, which can take years to complete and eat into corporate profits.
“Why bother with the time, bother, and expense of those pesky clinical trials to get your drug approved for additional indications, when you can rely on clinical experiences based on … uncontrolled observational studies, or registries instead?” Dr. Gorski wrote.
What’s more? Dr. Gorski isn’t even convinced this provision will help fulfill the Cures Act’s fundamental mission – to fix the FDA bottleneck and speed the approval of necessary medicines.
“If I were the CEO of a pharmaceutical company, I’d love it. Indeed, the one thing this provision most definitely does not do is to speed effective treatments to patients. Rather, it smacks of being a payoff to pharmaceutical companies,” he wrote.
Weak clinical trials already drive approvals
The fact is, however, that even now the FDA approves drugs and devices based on insufficient clinical data, usually with the caveat that the company must conduct postmarket trials to further support the safety and efficacy of their new product.
Oftentimes, those trials are not conducted in a manner from which any meaningful conclusions about product safety or efficacy can be derived.
A perspective published in the New England Journal of Medicine in 2015 highlights this problem, saying, “Most FDA-required postapproval studies are smaller than the premarketing studies, most follow patients for 1 year or less, and nearly half lack comparator groups.”
The NEJM perspective dove into the details of the clinical trials that led to the approval of Essure, a controversial permanent birth control device that made headlines and was the inspiration for two Congressional proposals: Ariel Grace’s Law and the Medical Device Guardians Act.
Essure was approved more than 14 years ago based on two nonrandomized, nonblinded, prospective studies that lacked a control group. The sterilization device is made of two metal coils implanted in a woman’s fallopian tubes. The coils incite an inflammatory reaction, which causes scar tissue to form around the coils and block the tubes.
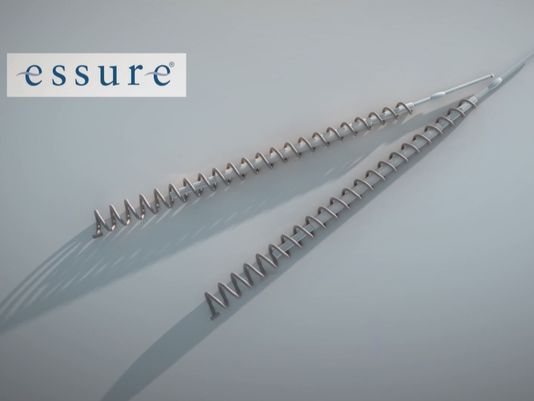 Essure is a permanent form of birth control, intended to remain inside a woman’s body for the rest of her life. Yet, clinical studies on Essure followed most of the participants for only one year following implantation and only 25% of participants for two years.
Essure is a permanent form of birth control, intended to remain inside a woman’s body for the rest of her life. Yet, clinical studies on Essure followed most of the participants for only one year following implantation and only 25% of participants for two years.
Since it came to market, the device has been used by more than 750,000 women worldwide, according to figures provided by Bayer Healthcare, the maker and most ardent defender of Essure.
Thousands of those women have reported serious and sometimes life-threatening side effects after getting Essure, like perforation of the uterus, fallopian tubes or other organs by the coils; migration of the coils from the tubes to other parts of the body; chronic and often debilitating pelvic pain; heavy and prolonged menstrual periods; allergic and autoimmune reactions; unintended pregnancies and ectopic pregnancies.
At least four women have died because of Essure and more than a dozen women lost pregnancies because of Essure, an analysis by the FDA found.
The concerns raised by thousands of women who reported a wide range of serious side effects with Essure could have been prevented or detected sooner, according to the NEJM perspective, “if there had been higher-quality premarketing and postmarketing evaluations.”
Rep. Mike Fitzpatrick, R-Pa., has been an advocate in Washington for those women suffering from Essure and another controversial medical device, the power morcellator, which is known to spread undetected cancers in some women undergoing minimally invasive hysterectomies.
The Congress member introduced Ariel Grace’s Law and the Medical Device Guardians Act to the House this year and attempted to tack them onto the 21st Century Cures Act.
Ariel Grace’s Law would allow patients harmed by class III medical devices, such as Essure, to file lawsuits against the manufacturers, thus removing the federal preemption protection now afforded to such devices.
The Medical Device Guardians Act would expand the current requirement for hospitals and manufacturers to report adverse events to the FDA to include doctors and doctor’s offices. The goal of the MDGA was to increase reporting of serious injuries and deaths from medical devices, which the FDA and numerous studies have found are vastly underreported, sometimes, unlawfully so.
Much to the dismay of Rep. Fitzpatrick, both bills failed to pass with the 21st Century Cures Act.
On the day of the House vote, the Congress member announced he would not vote for the Cures Act because the bill failed to provide enough protections for patients.
“I agree with many measures in this bill,” Rep. Fitzpatrick said in a press release. “However, I voted against the bill because it fails to protect patients against dangerous medical devices. For two years, I’ve sought medical device reform in Congress to raise awareness and advance legislation that protected patients and altered FDA processes and procedures to allow for maximum innovation and safety. Congress ignored the victims affected by faulty, dangerous medical devices …”
These women are now turning to the legal system to rectify the injustices they feel they were served by Bayer and the FDA.
Dozens of Essure lawsuits are pending in state and federal courts throughout the country, and many of them are gaining traction within those courts despite the federal preemption the device was granted upon its approval, which shields it from litigation.
Essure is merely one example of a dangerous medical device approved by the FDA based on weak clinical data and the further weakening of the FDA’s requirements could see many more slip through its cracks.
‘Breakthrough’ devices allowed fast-track approval
 Relaxing requirements for drug and device approval may serve to speed up the process, but it does nothing for patient safety. And this was just one of many provisions detailed in the Cures Act that would speed up approvals while leaving patient safety in the dust.
Relaxing requirements for drug and device approval may serve to speed up the process, but it does nothing for patient safety. And this was just one of many provisions detailed in the Cures Act that would speed up approvals while leaving patient safety in the dust.
The section “Breakthrough devices” seeks to expedite the approval process of any device that represents a breakthrough technology and would better treat life-threatening or debilitating conditions.
The consumer advocacy group Public Citizen warned in a summary of the bill that this category was “overly broad” and would put pressure on the FDA to rush approval of certain devices, which could lead to poor decisions.
The FDA already has an expedited approval process in place, its 510(k) clearance program, which lets companies submit for approval any medical device that is “substantially equivalent” to another device already on the market without conducting rigorous clinical tests to prove their device is safe and effective.
A “substantially equivalent” device is one that has the same intended use and has the same technological characteristics as a similar device. But devices using new technologies can still be submitted through 510(k) so long as it doesn’t “raise new questions of safety and effectiveness.”
If this process doesn’t raise the red flags in your head, it should. The many product liability lawsuits playing out in courtrooms across the country are centered on medical devices approved through the FDA’s 510(k) clearance program.
Product liability lawsuits demonstrate expedited approval shortcomings
Some of the most high-profile of these product liability lawsuits include the transvaginal mesh cases unfolding in a West Virginia district court. Tens of thousands of lawsuits alleging mesh manufacturers defectively designed their products and failed to warn about the potential risks were consolidated into multidistrict litigations (MDLs) in the early 2010s.
 Johnson & Johnson’s subsidiary Ethicon Inc., Boston Scientific, American Medical Systems, and C.R. Bard Inc. were all sued by women injured by the companies’ vaginal mesh products. Women have reported suffering terrible injuries, including adhesion of the mesh to surrounding tissues and perforation of nearby organs.
Johnson & Johnson’s subsidiary Ethicon Inc., Boston Scientific, American Medical Systems, and C.R. Bard Inc. were all sued by women injured by the companies’ vaginal mesh products. Women have reported suffering terrible injuries, including adhesion of the mesh to surrounding tissues and perforation of nearby organs.
The partners of these women have also claimed damages in these lawsuits due to another unintended consequence of the mesh – loss of consortium. That is, the mesh can make it too painful for women and their partners to have sex. There have been some reports from women who said their partners were injured by the mesh during sex because it had eroded through the vaginal wall.
Transvaginal mesh lawsuits have reaped millions of dollars in verdicts and settlements for victims, but there are still thousands of lawsuits awaiting trial.
Johnson & Johnson’s Ethicon division currently faces the most transvaginal mesh lawsuits of any mesh company – more than 31,000 in the West Virginia MDL alone. The device giant has been the most reluctant to settle, however, even after it was hit with a $12.5 million verdict out of Philadelphia in January.
The transvaginal mesh cases are just a drop in the bucket when it comes to harmful devices approved without rigorous testing.
Hernia meshes are another type of surgical mesh coming under fire for causing painful side effects and the need for additional surgeries. Just this month, more than a dozen federal lawsuits against Atrium Medical, the maker of a hernia mesh called C-Qur, were consolidated before a judge in New Hampshire.
Plaintiffs in the lawsuits allege suffering pain, inflammation, infection, mesh erosion and tissue damage as a result of the company’s mesh product. The FDA placed an injunction against the New Hampshire-based manufacturer and its parent company Maquet Holding in 2015, but Atrium chose to move some of its operations to another location instead of halting the production and sale of its hernia mesh.
Ethicon Inc. has also been sued over its hernia mesh product Physiomesh, which the company pulled from the shelves in 2014 after two large European studies showed the mesh was linked to greater rates of hernia recurrence compared to other meshes.
Establishing a “breakthrough” medical device category and urging the FDA to approve those devices even faster than the agency already is could lead to even more dangerous devices passing through the regulatory cracks undetected.
But the Cures Act isn’t limited to just medical devices; the act also works to erode critical FDA requirements governing approval of pharmaceutical drugs in this country.
‘Off-label’ promotion to insurance companies
 Under the 21st Century Cures Act, pharmaceutical companies can market their drugs for “off-label” uses to insurance companies without gaining FDA approval for that indication first.
Under the 21st Century Cures Act, pharmaceutical companies can market their drugs for “off-label” uses to insurance companies without gaining FDA approval for that indication first.
This means pharmaceutical companies can dramatically expand their markets while bypassing FDA approval requirements.
Many patients in this country have seen firsthand what can happen when a drug is prescribed “off-label” for a condition or patient population it was never approved to treat.
Patient injuries result of ‘off-label’ use
One high-profile example of damaging “off-label” use is in the case of the antipsychotic drug Risperdal, manufactured by Johnson & Johnson’s division Janssen Pharmaceuticals. The New Jersey-based company did not receive FDA approval to market its drug as a treatment for kids and teens until 2006 and 2007, but that did not stop the pharma giant from marketing the drug to doctors as a treatment for children long before its approval.
Used to treat behavioral and mental health disorders like autism, schizophrenia and bipolar 1, Risperdal is known to cause hormonal changes in children taking the drug. In young boys, the drug can cause breast tissue to grow abnormally large – a condition known as gynecomastia.
The company shelled out $2.2 billion to the Department of Justice in 2013 to settle accusations it illegally marketed Risperdal to treat children and elderly patients.
The company has also faced thousands of lawsuits brought by plaintiffs who took Risperdal and developed gynecomastia. Janssen has been hit with several million-dollar verdicts, including a $72 million verdict out of Philadelphia this past July.
 In the case of Risperdal, Janssen eventually gained approval for use of the drug in children, but in other cases, patients have been harmed by “off-label” uses of drugs that would never receive a nod from the FDA.
In the case of Risperdal, Janssen eventually gained approval for use of the drug in children, but in other cases, patients have been harmed by “off-label” uses of drugs that would never receive a nod from the FDA.
Zofran is a prominent example of this. The anti-nausea drug was approved by the FDA in the early 1990s to treat patients undergoing chemotherapy or surgery. But its maker, GlaxoSmithKline, found a way to expand its market share by targeting another group of people who often get nauseous – pregnant women.
The drug company began marketing Zofran as a way for pregnant women suffering from morning sickness to combat their queasiness – without adequately testing the drug to make sure it was safe for mother and her developing fetus.
Zofran has been linked to devastating birth defects, including holes in the heart and cleft lip and palate, among others.
GlaxoSmithKline settled a lawsuit with the U.S. government in 2012 for $3 billion – the largest settlement to date – over allegations the company illegally marketed some of its drugs, including Zofran, and failed to disclose safety data.
A series of lawsuits are underway now in courtrooms across the country brought by women who say they took Zofran during pregnancy and had babies born with birth defects as a result.
Is the 21st Century Cures Act Worth It?
 There’s no doubt that much of what the 21st Century Cures Act does in terms of funding and research is good for the American public and the U.S. healthcare system.
There’s no doubt that much of what the 21st Century Cures Act does in terms of funding and research is good for the American public and the U.S. healthcare system.
But at what cost?
The 21st Century Cures Act trades funding for deregulation in favor of Big Pharma. The act is already signed into law, which means these new deregulations will start to have profound effects on the U.S. healthcare system in due time.
Can Americans afford these erosions of essential regulations governing the safe passage of drugs and medical devices?
Considering the current state of affairs, the answer to that question is likely no.
Note: The information provided in this article is based on reports from publicly available sources, including news outlets, police reports, and eyewitness accounts. National Injury Help has not independently verified all details of the reported incident. If you find any inaccurate or outdated information, please contact us, and we will review and update the content as appropriate. The photo used in this post is for illustrative purposes only and does not depict the actual scene of the incident.
Disclaimer: The content of this article is intended for informational purposes only and does not constitute legal advice or establish an attorney-client relationship with National Injury Help. For legal assistance specific to your case, we encourage you to contact a qualified attorney.
Free Case Evaluation
Contact Us today for a FREE, Immediate Case Evaluation
Contact Us today for a FREE, Immediate Case Evaluation
Categories
Recent posts
- Why Helmet Law Disputes Can Complicate Motorcycle Injury Claims in California
- What Victims Need to Know When Dog Bite Injuries Exceed Insurance Policy Limits
- What Happens When an Out-of-State Driver Injures a Cyclist in California?
- The Legal Significance of Personality Changes After a Traumatic Brain Injury
- The Financial Burden of Long-Term Cognitive Rehabilitation After an Accident
What Our Clients Say